The Pathophysiology of Pernio-Like Lesions in COVID-19: A Synthesis of Virological, Thrombotic, and Immunological Mechanisms
“COVID toes” are caused by a strong local interferon response to SARS-CoV-2, leading to vascular inflammation and microthrombosis, despite mild symptoms and negative tests.

Executive Summary
The emergence of pernio-like lesions, colloquially termed "COVID toes," during the SARS-CoV-2 pandemic presented a clinical and pathophysiological paradox. These acral inflammatory lesions, characterized by erythema, edema, and sometimes vesicles, predominantly affect children and young adults who are otherwise healthy, often with mild or asymptomatic COVID-19 and consistently negative virological tests. This report provides an exhaustive analysis of the complex pathophysiology underlying this phenomenon, evaluating the evidence for three primary, interconnected hypotheses: direct viral damage to endothelial cells, microthrombosis in cutaneous small blood vessels, and a delayed, robust type I interferon (IFN-I) immune response.
The analysis concludes that the pathophysiology of "COVID toes" is not attributable to a single, isolated mechanism but rather a complex, sequential interplay of all three proposed pathways. The central and unifying driver is a potent, localized type I interferon response, likely initiated by a sparse and transient presence of SARS-CoV-2 viral components in the acral microvasculature. This robust innate immune reaction, while highly effective at achieving systemic viral clearance—thus explaining the mild clinical course and negative tests—induces a secondary, localized endotheliitis and a dense lymphocytic vasculitis. This IFN-I-driven inflammation damages the vascular endothelium, creating a prothrombotic microenvironment that leads to the formation of microthrombi. The resulting microvascular occlusion and localized ischemia produce the characteristic clinical signs of chilblains.
This unified model successfully reconciles the seemingly contradictory clinical observations. It reframes "COVID toes" not as a direct cytopathic effect of the virus, but as a form of "collateral damage" from an over-exuberant, yet systemically protective, innate immune response. The cutaneous pathology is the visible signature of a highly effective antiviral state. This understanding distinguishes the condition from the severe, systemic coagulopathy-driven acral ischemia seen in critically ill patients and highlights the critical role of host immune factors, and likely genetic predisposition, in determining the clinical manifestations of SARS-CoV-2 infection.
Introduction: Defining the Clinical and Histopathological Landscape of "COVID Toes"
The COVID-19 pandemic, caused by the novel coronavirus SARS-CoV-2, was primarily characterized by respiratory illness. However, it soon became apparent that the virus could induce a wide spectrum of extrapulmonary manifestations, among which cutaneous signs were prominent and varied.1 One of the most widely reported and debated of these was the sudden appearance of clusters of pernio-like lesions, or chilblains, particularly on the toes and fingers of younger individuals.1 These lesions, quickly dubbed "COVID toes" or "pseudo-chilblains," became a subject of intense medical and media attention, not only for their striking appearance but for the perplexing clinical context in which they arose.3 Understanding the clinical presentation, unique epidemiology, and underlying microscopic pathology of these lesions is fundamental to deciphering their complex etiology.
Clinical Presentation: Acral Erythema, Edema, and Vesicles
Clinically, "COVID toes" are largely indistinguishable from idiopathic perniosis (classic chilblains). The condition typically manifests as erythematous to violaceous or purpuric macules, papules, and nodules localized to acral sites.4 The toes are most commonly affected, but lesions can also appear on the fingers, heels, soles, and lateral aspects of the feet.7 The initial lesions are often bright red or pink before evolving to a deeper purple hue.7 In some cases, these lesions can progress to form vesicles, bullae, erosions, or ulcerations.3
Associated symptoms are variable. Patients may experience pain, pruritus (itching), or a burning sensation, while a significant portion of cases are entirely asymptomatic, with the discoloration being the only noticeable feature.4 The condition is typically self-limiting, with most lesions resolving spontaneously over a period of 10 days to several weeks without sequelae.1 However, a subset of individuals may experience a more protracted course, with persistent acrocyanosis or recurrent episodes of chilblains lasting for months, a phenomenon described as "long COVID toe".11
Epidemiological Context: The Paradox of Mild Disease and Negative Tests
The most enigmatic aspect of "COVID toes" is its epidemiology. The lesions appeared in distinct clusters that temporally coincided with peaks of the COVID-19 pandemic, often affecting children, adolescents, and young adults with no prior history of chilblains or relevant comorbidities like autoimmune disease or Raynaud's phenomenon.1 Crucially, the vast majority of these patients presented with either mild systemic symptoms of COVID-19 (e.g., low-grade fever, congestion) or were completely asymptomatic.1
This presentation created a central paradox that has fueled extensive debate: at the time the skin lesions appeared, most affected individuals tested negative for active SARS-CoV-2 infection via nasopharyngeal reverse transcriptase-polymerase chain reaction (RT-PCR) and also lacked a detectable antibody response (serology).5 This consistent clinical-virological disconnect led some researchers to question the causal relationship, suggesting that the increased incidence of chilblains could be an artifact of lifestyle changes during lockdowns (e.g., being barefoot on cold floors) or heightened reporting due to media attention.14 However, the prevailing view is that this paradox is not evidence against a causal link but is, in fact, a fundamental clue to the underlying pathophysiology. A highly efficient and rapid immune response could plausibly clear the virus from systemic circulation and mucosal sites before significant symptoms or a robust, measurable antibody response can develop. In this model, the localized skin inflammation is the only lingering sign of the host's successful encounter with the virus, reframing the "negative test" problem as a key piece of supporting evidence for an immune-mediated pathogenic mechanism.
The Microscopic View: A Characteristic Pattern of Lymphocytic Vasculitis
Histopathological examination of skin biopsies from "COVID toes" reveals a consistent and characteristic pattern of inflammation that provides crucial insights into the disease process. The hallmark microscopic feature is a dense, superficial and deep perivascular lymphocytic infiltrate.11 This inflammatory infiltrate is often notably concentrated around eccrine (sweat) glands, a finding known as perieccrine inflammation.10
Other common histopathological findings include significant edema in the papillary dermis, swelling and activation of endothelial cells lining the small blood vessels (a feature known as endotheliitis), and vacuolar changes at the dermo-epidermal junction.11 This constellation of features is diagnostic of a lymphocytic vasculitis. Importantly, this histological picture is virtually identical to that of idiopathic chilblains and the chilblains associated with autoimmune connective tissue diseases, particularly chilblain lupus erythematosus.16 This striking microscopic similarity strongly suggests a shared final common pathway of immune-mediated vascular injury, regardless of the initial trigger, and serves as a critical bridge to understanding the central role of specific immune pathways, such as the type I interferon response.
The Pathophysiological Triad: An Examination of Competing and Complementary Hypotheses
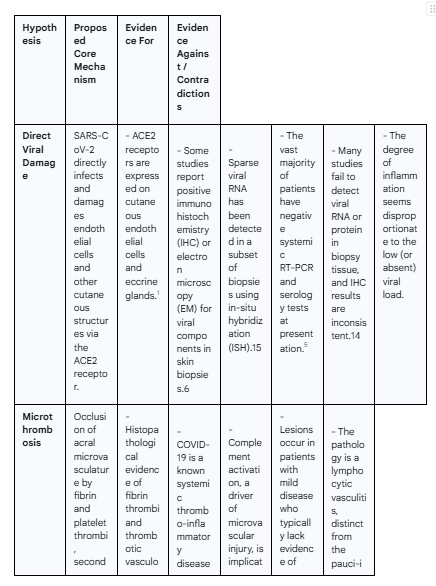
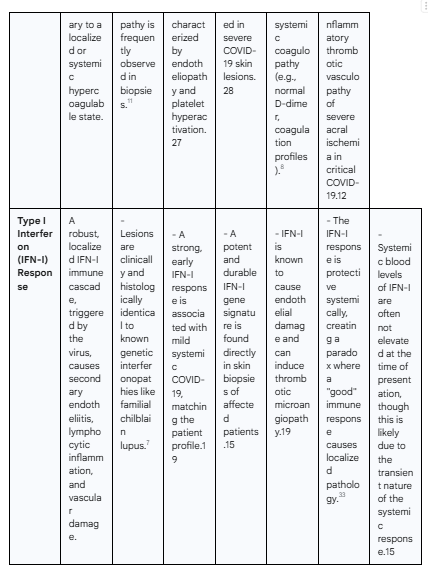
The investigation into the precise cause of "COVID toes" has centered on three primary mechanistic theories: direct viral damage to the cutaneous microvasculature, the formation of occlusive microthrombi, and a dysregulated, hyperactive type I interferon immune response. While initially considered as competing explanations, the accumulating evidence suggests these hypotheses are not mutually exclusive. Instead, they likely represent interconnected stages of a single, complex pathogenic cascade. The following table provides a structured overview of the evidence supporting and contradicting each hypothesis, setting the framework for a more detailed analysis.
(Editor’s note: Google still has problems exporting Gemini’s tables to Google Docs.)
Hypothesis I: Direct Viral Cytotoxicity and Endothelial Invasion
The most straightforward hypothesis for the pathogenesis of "COVID toes" posits that SARS-CoV-2 directly infects and damages the cells of the skin, particularly the endothelial cells lining the small blood vessels. This theory is grounded in the fundamental mechanism of viral entry and is supported by a subset of in-situ studies, though it is significantly challenged by a larger body of contradictory virological data.
The Biological Plausibility: ACE2 Receptors in Cutaneous Vasculature
For direct viral infection to occur, the target cells must express the necessary receptors for viral entry. The primary receptor for SARS-CoV-2 is angiotensin-converting enzyme 2 (ACE2), which works in concert with the transmembrane protease serine 2 (TMPRSS2) to facilitate viral fusion and entry.4 Research has confirmed that ACE2 is robustly expressed in various cutaneous structures. Specifically, ACE2 is present on the surface of endothelial cells of the cutaneous microvasculature, the smooth muscle cells of small vessels, and the epithelial cells of eccrine sweat glands.1 This distribution provides a direct and biologically plausible portal for SARS-CoV-2 to invade the very tissues that are pathologically affected in "COVID toes," lending strong initial support to the direct damage hypothesis. Viral binding to ACE2 can also disrupt the local renin-angiotensin-aldosterone system (RAAS), potentially leading to an increase in pro-inflammatory and vasoconstrictive angiotensin II, further contributing to vascular pathology.4
Evidence from In-Situ Viral Detection: A Review of IHC, ISH, and Electron Microscopy Findings
Several studies have sought to provide direct proof of this hypothesis by detecting the virus or its components within skin biopsy specimens. This evidence, while compelling in individual cases, has proven to be inconsistent across the literature.
Some of the most direct evidence comes from immunohistochemistry (IHC) studies, which use antibodies to stain for specific viral proteins in tissue sections. A number of reports have successfully demonstrated positive IHC staining for SARS-CoV-2 proteins, most notably the spike (S) and nucleocapsid (N) proteins, within the cytoplasm of endothelial cells and eccrine gland epithelium in biopsies from patients with pernio-like lesions.6 Further corroboration has come from a limited number of studies using electron microscopy (EM), which have visualized intracellular structures morphologically consistent with coronavirus particles within endothelial cells of small blood vessels and eccrine glands.8 More recently, highly sensitive molecular techniques like RNAscope, a form of in-situ hybridization (ISH), have been employed. One notable study from a Wisconsin cohort detected sparse SARS-CoV-2 viral RNA sequences in the toes of 20% of their patients and 33% of a validation cohort, suggesting that at least a subset of patients harbor viral genetic material in the affected tissue.15
The Case Against: Analyzing the High Rate of Negative Systemic and Local Viral Tests
Despite the affirmative findings in some studies, the direct viral damage hypothesis is substantially weakened by a large and consistent body of contradictory evidence. The primary challenge, as previously noted, is the overwhelming frequency of negative systemic tests (nasopharyngeal RT-PCR and serology) in patients at the time their skin lesions are present.5
More critically, many attempts to find the virus directly within the skin have failed. Multiple studies using RT-PCR and ISH on biopsy tissue from "COVID toes" have been unable to detect the viral genome, concluding that the lesions are unlikely to be actively infectious.14 The IHC data is also fraught with inconsistency. Some studies have reported staining for the spike protein but not the nucleocapsid protein, raising concerns about the specificity of the antibodies used and the potential for false-positive results due to cross-reactivity.14 A comprehensive study that analyzed skin biopsies from 52 patients with various COVID-19-associated cutaneous phenotypes found evidence of SARS-CoV-2 in only 38% of cases overall, with no clear association between viral presence and any specific clinical pattern, including pernio-like lesions.24 This suggests that even when the virus is present in the skin, it may not be the sole or primary driver of the observed pathology.
The Cleaved Spike Protein Theory: Endothelial Damage Without Viral Replication
A sophisticated theory has emerged to reconcile the conflicting findings of positive protein staining in the absence of detectable viral RNA. This hypothesis centers on the role of the viral spike protein itself as a pathogenic agent. One pivotal study found a clear discordance in biopsy samples: IHC staining for the spike protein was positive in endothelial cells, but RNA-ISH for the gene encoding the spike protein was negative in the exact same tissue sections.38
This finding suggests that cleaved, circulating fragments of the spike protein, potentially part of incomplete viral particles or "pseudovirions," may be sufficient to cause pathology.30 These fragments could be shed from a distant site of infection (like the respiratory tract), travel through the bloodstream, and bind to ACE2 receptors on acral endothelial cells without the need for active, local viral replication.30 The binding of the spike protein alone is known to be capable of triggering endothelial cell activation, inflammation, and complement deposition. This model elegantly explains how endothelial damage can occur and why viral proteins might be detected in the absence of the full viral genome. It reframes the role of the virus from that of a sustained, cytopathic agent to that of a transient, potent "trigger" or pathogen-associated molecular pattern (PAMP). This initial interaction acts as the spark that ignites a much larger, host-driven inflammatory fire, which then becomes the primary driver of the disease process.
Hypothesis II: Microthrombosis and Complement-Mediated Vasculopathy
A second major hypothesis proposes that "COVID toes" are a manifestation of microvascular thrombosis, where small blood clots form and occlude the capillaries of the skin. This theory places the lesions within the broader context of COVID-19 as a systemic disease characterized by endothelial dysfunction and a prothrombotic state. While evidence of thrombosis is clear, its role appears to be secondary to an inflammatory process rather than a primary coagulopathy in these specific patients.
Histopathological Evidence of Thrombotic Occlusion in Pernio-like Lesions
Direct microscopic examination of biopsy specimens provides the most compelling evidence for this hypothesis. Numerous histopathology reports on "COVID toes" have documented the presence of fibrin thrombi partially or completely occluding the lumina of small blood vessels and capillaries within the papillary dermis.11 Studies using specialized stains have confirmed these occlusions are composed of fibrin.11 One detailed case report of a patient with persistent "long COVID toe" demonstrated ongoing thrombotic vasculopathy with endothelial swelling and capillary thrombosis 16 weeks after the presumed initial infection, highlighting the potential for durable vascular injury.11 These findings unequivocally confirm that microthrombosis is a component of the pathology.
Connecting "COVID Toes" to Systemic COVID-19 Coagulopathy
The presence of localized thrombi in the toes is mechanistically linked to the well-established understanding that severe COVID-19 is a systemic thrombo-inflammatory disease.1 The pathogenesis of this coagulopathy is multifactorial, involving widespread endothelial injury and inflammation (endotheliopathy), hyperactivation of platelets, and dysregulation of the coagulation cascade.27 Activated endothelial cells and platelets release highly procoagulant microvesicles (MVs), which are found at elevated levels in COVID-19 patients and correlate with disease severity and markers of thrombosis like D-dimer.29 This systemic prothrombotic environment provides a plausible backdrop for the formation of microthrombi in the distal microvasculature of the toes.
The Role of Complement Activation
A key driver of COVID-19-associated microvascular injury is the dysregulated activation of the complement system, a critical part of innate immunity. In severe COVID-19, particularly in patients with severe skin manifestations like retiform purpura, biopsies show extensive deposition of complement components, such as C4d and the C5b-9 membrane attack complex, within the microvasculature.28 This indicates a process of complement-mediated thrombotic microangiopathy, where complement activation on the endothelial surface triggers inflammation, platelet aggregation, and clot formation.28 While this has been most clearly demonstrated in severe disease, it is likely that a similar, albeit more localized and less intense, complement-mediated process contributes to the endothelial injury and microthrombosis seen in pernio-like lesions.
Distinguishing Pernio-like Lesions from Severe Acral Ischemia
A crucial point of clarification is the fundamental difference between the microthrombosis observed in "COVID toes" and the severe, often necrotic, acral ischemia seen in critically ill, hospitalized COVID-19 patients.12 Although both involve vascular occlusion, their underlying pathophysiology and clinical implications are distinct.
Severe acral ischemia is a direct manifestation of systemic hypercoagulability, occurring in patients with markedly elevated D-dimer levels, disseminated intravascular coagulation (DIC), and other signs of a systemic prothrombotic state. Histologically, it is often a pauci-inflammatory thrombotic vasculopathy, and it portends a very poor prognosis.12 In stark contrast, "COVID toes" occur in patients with mild disease who typically have normal coagulation profiles and D-dimer levels.8 The histopathology is not one of bland thrombosis but of a florid lymphocytic vasculitis, where the microthrombi appear to be a secondary consequence of the intense vessel wall inflammation. This distinction is critical: the type of vascular occlusion reflects the underlying immune status of the host. In patients with a robust and effective immune response (mild disease), the pathology is an inflammatory vasculitis with secondary microthrombi. In patients with a failed immune response and a subsequent cytokine storm (severe disease), the pathology is a primary, complement-driven thrombotic vasculopathy. Therefore, the thrombus in "COVID toes" is an endpoint of an immune-driven process, not the primary event itself.
Hypothesis III: The Type I Interferon Signature as a Central Driver
The most compelling and unifying hypothesis for the pathogenesis of "COVID toes" centers on the role of the type I interferon (IFN-I) system. This theory posits that the skin lesions are a paradoxical manifestation of a highly effective, robust antiviral immune response. In this model, the IFN-I response is both the protector against severe systemic disease and the direct instigator of the localized cutaneous pathology.
The IFN-I Response: Protector in Systemic Disease, Pathogen in the Skin
Type I interferons (primarily IFN-α and IFN-β) are a family of cytokines that form the first line of defense against viral infections.12 A strong, early systemic IFN-I response is critical for controlling SARS-CoV-2 replication, preventing widespread tissue damage, and orchestrating an effective adaptive immune response.12 Abundant evidence shows that patients with mild or asymptomatic COVID-19 mount such a response, leading to rapid viral clearance.12 Conversely, a hallmark of severe, life-threatening COVID-19 is a delayed, attenuated, or otherwise impaired IFN-I response, which allows for uncontrolled viral replication and a subsequent, damaging hyperinflammatory state or "cytokine storm".33
The "COVID toes" phenomenon fits perfectly into this paradigm. The lesions occur almost exclusively in patients with the mild disease course characteristic of a successful IFN-I response. The hypothesis is, therefore, that "COVID toes" are the direct, localized consequence—a form of "collateral damage"—of this systemically protective immune state.12 The very immune mechanism that saves the patient from severe lung disease is the same one that attacks the microvasculature of their skin.
Parallels with Genetic Interferonopathies: Lessons from Familial Chilblain Lupus
Perhaps the most powerful line of evidence supporting the IFN-I hypothesis is the striking resemblance of "COVID toes" to a group of rare genetic disorders known as type I interferonopathies. Conditions like Familial Chilblain Lupus (FCL), caused by mutations in genes such as TREX1, are defined by the chronic, constitutive overproduction of IFN-I.7 The defining clinical feature of these genetic diseases is the development of severe, recurrent chilblains that are clinically and histologically identical to "COVID toes".15
This provides an unequivocal precedent: a systemically elevated IFN-I state is known to be sufficient to cause this specific pattern of acral vasculopathy. This parallel allows for the reconceptualization of "COVID toes" as a transient, virally-triggered, acquired interferonopathy. In the genetic form, the IFN-I overproduction is permanent and hard-wired. In "COVID toes," a viral trigger initiates a massive but ultimately self-limited burst of IFN-I. This temporary burst is sufficient to induce the same cutaneous pathology seen in the chronic genetic forms, elegantly explaining why the lesions appear, why they look exactly like lupus chilblains, and why they eventually resolve in most patients.
Immunological Evidence: Measuring IFN-I Signatures in Blood and Skin Biopsies
This hypothesis is not merely based on analogy; it is supported by direct immunological measurements in patients with "COVID toes."
In Skin: Multiple studies have demonstrated a potent and durable IFN-I signature directly within the affected skin tissue. Biopsies of "COVID toes" show significantly elevated local expression of IFN-stimulated genes (ISGs), such as Myxovirus resistance protein A (MxA).32 Furthermore, the inflammatory infiltrate is rich in plasmacytoid dendritic cells (pDCs), which are the body's most potent producers of IFN-I, confirming that the machinery for IFN-I production is present and active at the site of injury.15 This local activation of the IFN-I pathway is significantly higher than in normal skin.15
In Blood: The systemic IFN-I signature appears to be highly transient. Studies in humans and animal models show that the IFN-I response in the peripheral blood peaks very early after viral exposure and then wanes within days.15 This finding is critical as it resolves an apparent contradiction in the data. Blood samples are typically drawn when patients present with skin lesions, which can be weeks after the initial, often asymptomatic, infection. By this time, the systemic IFN-I response has returned to baseline, explaining why many studies find normal IFN levels in the blood. However, the signature remains durably elevated within the skin tissue itself, driving the ongoing pathology.15
Mechanism of IFN-I-Mediated Endothelial Damage and Inflammation
The final piece of the puzzle is understanding how high local concentrations of IFN-I lead to the observed vascular damage. IFN-I exerts several pathogenic effects on the microvasculature. It promotes a potent pro-inflammatory state by upregulating chemokines that recruit cytotoxic lymphocytes (T cells and NK cells) to the vessel wall, leading to the characteristic lymphocytic vasculitis.35 It directly activates endothelial cells, causing them to swell, become dysfunctional (endotheliitis), and express adhesion molecules that further amplify the inflammatory infiltrate.35
Crucially, the IFN-I response directly links to the other pathogenic hypotheses. IFN-I is known to create a prothrombotic state on the endothelial surface. Indeed, recombinant IFN-I therapy has been reported to induce thrombotic microangiopathy in some patients, providing a direct causal link between the IFN-I response and the microthrombi observed in biopsies.19 Furthermore, IFN-I can promote complement-mediated endothelial injury, tying it to the mechanisms of complement activation seen in more severe COVID-19.35 Thus, the IFN-I response acts as the central hub, initiating the inflammation that leads to secondary thrombosis and vascular occlusion.
Synthesis: A Unified Pathophysiological Model for COVID Toes
The evidence reviewed does not support one hypothesis to the exclusion of the others. Instead, it points toward a unified, sequential model where a viral trigger initiates a potent immune cascade that culminates in thrombotic vascular injury. This integrated framework provides the most comprehensive explanation for the clinical, virological, and histopathological features of "COVID toes."
Step 1: Viral Trigger and Initial Endothelial Interaction
The cascade begins with the arrival of SARS-CoV-2 components in the distal microvasculature of the acral skin. This may involve intact virions, but given the difficulty in detecting viral RNA, it is more likely to involve sparse, circulating fragments of the virus, particularly the cleaved spike protein.30 These viral components bind to ACE2 receptors expressed on the surface of endothelial cells and eccrine glands.1 This interaction, representing the direct viral involvement of Hypothesis I, is likely minimal and transient but serves as the critical PAMP to alert the innate immune system.
Step 2: A Robust, Localized Type I Interferon Cascade
In individuals with a predisposition for a strong innate immune response—a trait that may be genetically determined by variations in pathways involving Toll-like receptor 7 (TLR7) or other pattern recognition receptors—this minimal viral trigger is met with an exceptionally potent response.35 Local pDCs and other immune cells recognize the viral components and unleash a massive and sustained local production of type I interferons (Hypothesis III).15 This response is highly effective at neutralizing any local viral threat, preventing systemic viral replication and spread. This explains the mild or absent systemic symptoms and the subsequent negative RT-PCR and serology tests.
Step 3: IFN-I-Mediated Endotheliitis and Lymphocytic Vasculitis
The sustained high concentration of IFN-I in the cutaneous microenvironment becomes the primary pathogenic driver. It acts as a powerful chemoattractant, recruiting a dense infiltrate of cytotoxic T-lymphocytes and NK cells to the small blood vessels. Simultaneously, it directly activates the endothelial cells, causing swelling, dysfunction, and the upregulation of inflammatory mediators. The result is the hallmark histopathology of "COVID toes": a florid lymphocytic vasculitis with prominent endotheliitis. This is the core inflammatory event responsible for the tissue damage.
Step 4: Secondary Microthrombosis and Ischemic Manifestation
The inflamed, damaged, and dysfunctional endothelium loses its natural anticoagulant properties and becomes prothrombotic. The inflammatory milieu, potentially coupled with localized complement activation, promotes platelet aggregation and the formation of fibrin microthrombi within the capillaries (Hypothesis II).11 This vascular occlusion impedes blood flow, leading to localized ischemia and hypoxia. This final ischemic step produces the clinical manifestations of chilblains: erythema and violaceous discoloration from vascular stasis and deoxygenation, edema from vascular leakage, and pain or pruritus from ischemic nerve irritation.
Explaining the Clinical Paradox
This unified model elegantly resolves the central paradox of "COVID toes." It explains how a sign of pathology can be associated with a favorable systemic prognosis. The toe lesion is not caused by the failure to control the virus, but rather by the over-exuberant success of the immune system in a specific tissue compartment. The patient is systemically protected because of the strong IFN-I response that rapidly clears the virus. The "COVID toe" is the visible, localized price paid for that highly effective systemic protection.
Conclusion: Unresolved Questions and Future Research Directions
The pathophysiology of pernio-like lesions in COVID-19 is best understood not as a result of a single cause, but as the outcome of a pathogenic cascade. The most plausible model posits that a sparse viral trigger in the acral microvasculature initiates a robust, localized type I interferon response. This potent immune reaction, while systemically protective, induces a secondary lymphocytic vasculitis and endotheliitis, which in turn creates a prothrombotic state leading to microvascular occlusion and the clinical signs of chilblains. This model successfully integrates the virological, thrombotic, and immunological evidence and explains the paradoxical presentation of cutaneous pathology in the context of mild systemic disease.
Despite this progress, several key questions remain, highlighting important avenues for future research:
Genetic Predisposition: The development of "COVID toes" in only a subset of individuals with mild infection strongly suggests a host predisposition. Future research should focus on identifying specific genetic variants, particularly in genes controlling innate immune pathways like TLR7, TREX1, and other components of the IFN-I signaling cascade, that may prime individuals for this hyper-IFN-I response.35
Long-Term Sequelae: While most cases resolve completely, a notable subset of patients experiences persistent or recurrent symptoms, a condition termed "long COVID toe".11 The mechanisms driving this chronicity are unknown. Longitudinal studies are needed to understand the risk factors for this outcome and to determine if it involves persistent vascular damage, ongoing low-grade inflammation, or the development of autoimmunity.
The Role of Other Triggers: The reported increase in chilblains during the pandemic was not exclusively in patients with a clear link to SARS-CoV-2.17 It is possible that the same IFN-mediated vasculopathic pathway can be triggered by other stimuli, including other seasonal respiratory viruses or even non-infectious environmental factors associated with pandemic-related lifestyle changes.35 Delineating the full range of potential triggers for this pathology is an important area of investigation.
From a clinical perspective, these findings have direct implications. "COVID toes" should be recognized by clinicians as a potential sign of a recent, well-controlled encounter with SARS-CoV-2, even in the face of negative standard virological tests. Management should remain largely supportive, focusing on symptomatic relief and gentle warming. The pathophysiology, being primarily inflammatory rather than a systemic coagulopathy, argues against the routine use of anticoagulants unless there is separate evidence of a systemic thrombotic process. For severe or persistent cases, future therapeutic strategies might consider targeting the IFN pathway or using vasodilators, though further research is required to establish the safety and efficacy of such interventions. Ultimately, "COVID toes" serve as a remarkable clinical model of the intricate and often paradoxical relationship between viral infection, host immunity, and tissue-specific pathology.
The Cellular Energetics Crisis in Long COVID: A Mechanistic Analysis of SARS-CoV-2-Induced Mitochondrial Dysfunction in Skeletal Muscle
Long COVID causes mitochondrial collapse via viral proteins, immune dysregulation, and microvascular damage—triggering fatigue, muscle injury, and post-exertional malaise.
Sep 09, 2025
Executive Summary
The post-acute sequelae of SARS-CoV-2 infection (PASC), commonly known as Long COVID, presents a formidable global health challenge, characterized by a constellation of persistent, debilitating symptoms. Among these, profound fatigue, muscle weakness, and post-exertional malaise (PEM) are particularly prevalent and disabling, pointing towards a fundamental disruption in systemic energy metabolism.1 An accumulating body of evidence now converges on mitochondrial dysfunction within skeletal muscle as a central pathophysiological hub of the disease.1 This report provides a comprehensive mechanistic analysis of how SARS-CoV-2 infection initiates and perpetuates long-term mitochondrial impairment in skeletal muscle cells. The pathology is not the result of a single insult but rather a complex, multifactorial process driven by a "triad of insult": (1) direct molecular disruption by persistent viral components, particularly the Spike protein, which can directly interfere with mitochondrial respiratory machinery; (2) sustained collateral damage from a dysregulated immune response and chronic, low-grade inflammation that transcriptionally suppresses mitochondrial energy production pathways; and (3) a profound cellular energy crisis precipitated by widespread endothelial dysfunction and vascular microclotting, which leads to critical tissue hypoxia.
These converging pathways cripple the capacity of skeletal muscle for aerobic energy production, evidenced by diminished oxidative phosphorylation (OXPHOS) capacity, impaired electron transport chain (ETC) function, and a forced reliance on inefficient anaerobic glycolysis.4 This bioenergetic failure is accompanied by significant structural damage to the muscle tissue itself, including myofiber atrophy, a pathological shift towards highly fatigable fiber types, and exercise-induced necrosis.4 The result is a state of extreme bioenergetic fragility, where even minor increases in energy demand from physical or cognitive exertion can trigger a cascade of further metabolic and structural damage. This cascade is the biological basis of PEM, which clinically manifests as a delayed and prolonged exacerbation of symptoms.6 Understanding these intricate molecular mechanisms is paramount for developing targeted diagnostics and effective therapeutic interventions for the millions affected by this condition.
Section 1: The Pathological Milieu of Skeletal Muscle in Long COVID
Before delving into the molecular drivers of mitochondrial failure, it is essential to characterize the tissue-level consequences of these insults. Skeletal muscle in individuals with Long COVID is not merely deconditioned due to prolonged illness and inactivity; rather, it exhibits a distinct and active pathology. Histopathological, metabolic, and functional analyses of muscle tissue reveal a state of chronic injury, maladaptation, and extreme vulnerability to metabolic stress, providing a clear biological foundation for the profound fatigue and exercise intolerance that define the clinical presentation of the disease.
1.1 Histopathological Evidence: Myofiber Atrophy, Fiber-Type Shifting, and Inflammation
Direct examination of skeletal muscle biopsies from patients with Long COVID reveals a consistent pattern of structural abnormalities indicative of ongoing muscle damage and dysregulation.5 One of the most prominent features is myofiber atrophy, with an abundance of very small, angulated muscle fibers observed in patient samples.4 This reduction in muscle fiber size contributes to the overall muscle wasting, weakness, and decline in physical performance reported by patients.8 Concurrently, there are signs of an active, albeit potentially inefficient, repair process, evidenced by a significantly higher number of muscle fibers with internal nuclei, a marker of regeneration.4 These histopathological findings, which also include myofibrillar disorganization and signs of sarcolemmal damage, confirm that the muscle is in a state of persistent injury and attempted remodeling.10
Beyond simple atrophy, a critical maladaptation occurs at the level of muscle fiber composition. Biopsies from Long COVID patients show a significant shift towards a higher proportion of glycolytic, fast-twitch Type IIx fibers compared to the more mitochondria-rich, oxidative fibers seen in healthy, active controls.4 These Type IIx fibers are characterized by having fewer mitochondria and a lower density of surrounding capillaries, rendering them highly fatigable and poorly suited for sustained aerobic activity.11 This structural adaptation fundamentally lowers the muscle's intrinsic capacity for oxygen-dependent energy production, predisposing it to rapid exhaustion and a reliance on inefficient anaerobic metabolism. The convergence of these findings—structural maladaptation, persistent inflammation, and ongoing repair—paints a clear picture of skeletal muscle that is not merely deconditioned but is actively pathological. This altered architecture is a logical, albeit detrimental, response to a systemic environment where oxygen delivery or mitochondrial utilization is chronically impaired, setting the stage for the profound energy crisis that manifests as PEM.
This pathological environment is further characterized by persistent, low-grade inflammation. Immunohistochemical analysis of muscle tissue has identified the infiltration of immune cells, including CD3+ T-cells and a greater presence of CD68+ macrophages, indicating an ongoing, localized immune response within the muscle itself.4 This chronic inflammatory state can directly contribute to muscle dysfunction by promoting protein degradation, inhibiting protein synthesis, and generating oxidative stress, further exacerbating the cycle of tissue damage and impaired function.8
1.2 The Signature of Exertion: Exercise-Induced Myopathy and the Biological Basis of PEM
Post-exertional malaise (PEM) is the cardinal symptom of Long COVID and related post-infectious illnesses, defined as the worsening of symptoms following physical, cognitive, or emotional exertion.7 Far from being a subjective experience of fatigue, compelling evidence from longitudinal biopsy studies demonstrates that PEM has a direct and measurable biological correlate of acute tissue injury.6
In a landmark study, researchers performed skeletal muscle biopsies on Long COVID patients with PEM and healthy controls both before and one day after a single session of maximal cycling exercise.4 While the healthy controls showed minimal changes, a staggering 36% of the Long COVID patients exhibited large areas of necrotic, or dead, muscle fibers in their post-exercise biopsies.4 This severe, exercise-induced myopathy was accompanied by a worsening of local and systemic metabolic disturbances and an increase in tissue deposits.6 These findings provide unequivocal evidence that exertion in this patient population can trigger a cascade of events leading to acute cellular death and a worsening of the underlying pathology. This critical observation reframes PEM from a symptom to be managed into a harmful biological process to be avoided. It fundamentally challenges therapeutic approaches like graded exercise therapy, which could be iatrogenic, and provides a strong biological rationale for energy management strategies such as pacing to prevent the repeated induction of tissue injury.7
1.3 The Presence of Anomalous Deposits: Amyloid and Immune Infiltrates
A further pathological feature identified in the skeletal muscle of Long COVID patients is the presence of anomalous protein aggregates. Biopsies have revealed a significantly higher concentration of amyloid-containing deposits in the muscle tissue of patients compared to controls.4 These deposits are located in the extracellular matrix between muscle fibers and adjacent to endothelial cells lining the capillaries.6 Critically, the concentration of these amyloid deposits increases in patients following the same bout of exhaustive exercise that induces muscle necrosis.4 While the exact composition and pathological role of these deposits are still under investigation, their presence and exercise-induced accumulation suggest they may contribute to the local inflammatory environment, impair microcirculation, or interfere with nutrient and oxygen exchange between capillaries and muscle fibers, further compounding the tissue's metabolic distress.
Section 2: The Triad of Insult: Primary Drivers of Mitochondrial Injury
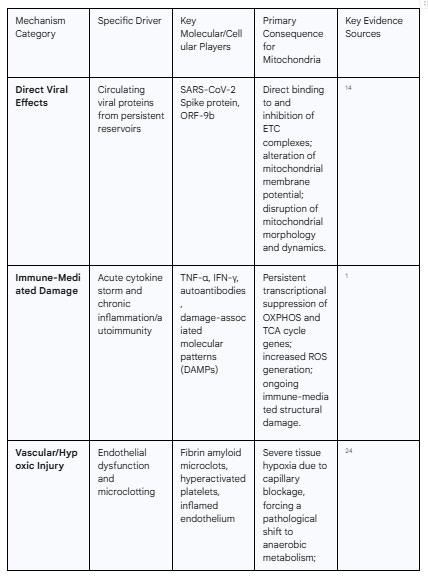
The profound muscle pathology observed in Long COVID is the downstream result of a multi-pronged assault on cellular energy systems initiated by the SARS-CoV-2 virus. The evidence points not to a single mechanism but to a "triad of insult," where direct viral interference, dysregulated immune responses, and severe vascular compromise converge to damage mitochondria. These three pathways are not mutually exclusive; they often overlap and amplify one another, creating a self-perpetuating cycle of cellular injury that persists long after the acute infection has resolved.
2.1 Direct Viral Hijacking of Mitochondrial Machinery
While persistent viral replication within skeletal muscle tissue itself has not been consistently demonstrated in Long COVID patients 5, a substantial body of evidence indicates that SARS-CoV-2 viral components can directly target and disrupt mitochondrial function. This suggests that the primary pathology may not require a chronic, localized infection. Instead, circulating viral proteins, potentially shed from persistent viral reservoirs in other tissues like the gut or nervous system, may be sufficient to drive ongoing mitochondrial damage throughout the body.14
In vitro studies have been instrumental in elucidating these direct mechanisms. Research has shown that various SARS-CoV-2 proteins, including the Spike protein and non-structural proteins encoded by Open Reading Frames (ORFs) such as ORF-9b, can translocate into the host cell's mitochondria and reside within the mitochondrial matrix.15 Once inside, these viral proteins can act as molecular saboteurs. The Spike protein, for instance, has been shown to directly interact with and impair the enzymatic activities of the electron transport chain (ETC) complexes, which are the core machinery of aerobic energy production.17 This interference leads to a measurable decrease in mitochondrial oxygen consumption rates, a reduction in the mitochondrial membrane potential essential for ATP synthesis, and visible structural damage, including mitochondrial fragmentation and swelling.17 Other viral proteins can bind to mitochondrial complexes, disrupting critical functions like calcium homeostasis, which can trigger cell death pathways.15 The concept of a distant viral reservoir providing these circulating proteins is supported by studies that have detected SARS-CoV-2 proteins in the blood of Long COVID patients up to 14 months after their acute infection, with a higher likelihood of detection in those with multisystem symptoms.14 This model, centered on systemic exposure to toxic viral components rather than localized muscle infection, better aligns with the widespread, multi-organ nature of Long COVID.
2.2 The Cascade of Immune-Mediated Damage
The host's own immune response to SARS-CoV-2 is a second, powerful driver of long-term mitochondrial dysfunction. The "cytokine storm" characteristic of acute COVID-19 can leave a lasting, detrimental imprint on cellular metabolism, a form of "molecular memory" that persists long after the virus is cleared. A longitudinal study using a hamster model of respiratory SARS-CoV-2 infection provided crucial insights into this process. The study found that while the virus did not directly invade skeletal muscle, the acute infection triggered a profound and persistent downregulation of nuclear genes essential for mitochondrial function. This transcriptional suppression affected genes involved in fatty acid β-oxidation (a key fuel source for muscle), the tricarboxylic acid (TCA) cycle, and all five protein complexes of the oxidative phosphorylation (OXPHOS) system, with the suppression lasting for at least 60 days post-infection.5
Further investigation revealed that this long-term suppression was likely initiated by a specific cytokine signature during the acute phase. In vitro experiments on muscle cell lines demonstrated that a synergistic combination of interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α)—but not either cytokine alone—was sufficient to markedly impair mitochondrial respiration and force a pathological shift towards anaerobic glycolysis.5 This suggests that the initial immune event can act as a molecular switch, triggering a self-perpetuating cellular program of mitochondrial suppression that explains the persistence of dysfunction even when systemic inflammatory markers have returned to normal.
This immune dysregulation can become chronic in Long COVID, transitioning into a state of persistent, low-grade inflammation.21 This creates a vicious cycle where mitochondrial dysfunction and inflammation fuel each other. Damaged mitochondria release their internal components, such as oxidized mitochondrial DNA (mtDNA) and cardiolipin, into the cytoplasm. The cell recognizes these molecules as damage-associated molecular patterns (DAMPs), which in turn activate inflammasomes like NLRP3, leading to the production of pro-inflammatory cytokines and perpetuating both local and systemic inflammation.1 Furthermore, there is growing evidence that the initial infection can trigger an autoimmune response, leading to the production of autoantibodies that mistakenly target the body's own mitochondrial proteins. This potential for continuous, immune-mediated damage to mitochondria provides another mechanism for the chronicity of the condition.1
2.3 Vascular Compromise and Tissue Hypoxia
The third major driver of mitochondrial injury is a profound and widespread pathology of the microvasculature. This mechanism acts as a powerful amplifier and perpetuator of mitochondrial dysfunction by creating a "supply chain crisis" that starves tissues of the oxygen essential for aerobic energy production. A central element of this vascular pathology is the formation of persistent, anomalous fibrin amyloid microclots.23 The SARS-CoV-2 Spike protein has been shown to induce a conformational change in fibrinogen, causing it to polymerize into an amyloid-like structure that is highly resistant to the body's natural clot-dissolving (fibrinolytic) processes.24
These persistent microclots, along with hyperactivated platelets, malformed blood cells, and inflammation of the capillary lining (endothelialitis), can physically obstruct the microcirculation.23 This blockage severely limits the passage of red blood cells through the capillaries, resulting in a dramatic reduction in oxygen delivery and profound tissue hypoxia in downstream organs, including the highly metabolic skeletal muscle.24 This state of chronic malperfusion can also lead to damaging cycles of ischemia (lack of blood flow) followed by reperfusion (restoration of blood flow), a process known to generate massive bursts of oxidative stress that further damage mitochondria.25
The microclot and hypoxia hypothesis is strongly corroborated by data from invasive cardiopulmonary exercise testing (iCPET). These studies, which directly measure physiological parameters during exercise, have consistently shown that a primary mechanism of exercise limitation in Long COVID patients is severely impaired systemic oxygen extraction.26 This means that even when oxygen is present in the blood, the tissues—particularly skeletal muscle—are unable to effectively extract and utilize it. This finding points directly to a problem at the interface between the capillaries and the muscle cells, a bottleneck fully consistent with microvascular obstruction and a critical lack of oxygen supply to the mitochondria.27 This chronic hypoxia forces the muscle cells into a state of "pseudo-anaerobiosis," compelling them to rely on inefficient anaerobic glycolysis even at low levels of exertion, providing a direct link between the systemic vascular pathology and the specific cellular metabolic crisis observed in the muscle.

Section 3: The Molecular Signature of Mitochondrial Collapse
The convergence of direct viral effects, immune-mediated damage, and vascular-induced hypoxia unleashes a catastrophic breakdown of mitochondrial function at the molecular level. This is not a single-point failure but a comprehensive systemic collapse of the organelle's ability to produce energy, manage oxidative stress, and maintain its own structural integrity. The molecular signature of this collapse provides a granular explanation for the profound bioenergetic deficits observed in Long COVID.
3.1 Catastrophic Failure of Energy Production: Impaired OXPHOS, TCA Cycle, and ETC Function
The core function of mitochondria is to generate adenosine triphosphate (ATP), the cell's energy currency, through the highly efficient process of oxidative phosphorylation (OXPHOS). In Long COVID, this central pathway is severely compromised. Direct measurements in skeletal muscle tissue from patients reveal a significantly lower OXPHOS capacity compared to healthy controls.4 This fundamental deficit in the ability to produce energy aerobically is not a transient issue; it is a persistent state that remains low at rest and, critically, fails to augment—and may even worsen—following exertion.4
This failure is evident at multiple stages of the energy production pipeline. Transcriptional analyses show a sustained downregulation of the nuclear genes that encode the protein machinery for both the tricarboxylic acid (TCA) cycle and all five complexes of the electron transport chain (ETC).5 The TCA cycle is the metabolic engine that processes fuel molecules to generate the high-energy electrons (NADH and FADH2) needed by the ETC. Its suppression means that the primary fuel supply for aerobic respiration is choked off. Metabolomic studies confirm this, showing a depletion of key TCA cycle intermediates like alpha-ketoglutarate and citric acid in patient muscle, alongside an accumulation of metabolites associated with anaerobic glycolysis.4 This metabolic profile is the clear signature of a system that has abandoned its primary aerobic pathway in favor of a less efficient anaerobic backup.
The ETC itself is also directly impaired. The activity of succinate dehydrogenase (SDH), which functions as Complex II of the ETC as well as a key enzyme in the TCA cycle, is a crucial indicator of mitochondrial function. While baseline SDH activity may be comparable to controls, it is significantly reduced in Long COVID patients in the 24 hours following the induction of PEM.4 This finding is particularly revealing, as it suggests that the mitochondrial population is not only dysfunctional but also fragile and unable to withstand metabolic stress. The very act of trying to produce more energy appears to damage or deplete the existing mitochondrial machinery, providing a direct molecular explanation for the post-exertional "crash."
The dysfunction culminates at the final stage of ATP synthesis. Studies on peripheral blood mononuclear cells (PBMCs) from Long COVID patients have uncovered a remarkable abnormality in ATP synthase (Complex V). In a state of severe bioenergetic distress, this enzyme, which normally uses the proton gradient across the inner mitochondrial membrane to synthesize ATP, can run in reverse. It begins to hydrolyze (break down) ATP to pump protons back across the membrane, sacrificing energy to maintain the critical mitochondrial membrane potential.31 This reversal is a profound indicator of a system on the brink of collapse, where the cell is actively consuming its limited energy reserves just to prevent catastrophic failure of the organelle. The combination of impaired fuel processing, a broken ETC, and a reversed ATP synthase constitutes a complete system failure of aerobic energy production.
3.2 The Vicious Cycle of Oxidative Stress
Mitochondrial dysfunction and oxidative stress are inextricably linked in a self-perpetuating vicious cycle that drives progressive cellular damage.1 A healthy ETC carefully passes high-energy electrons down a chain of protein complexes, using their energy to generate the proton gradient for ATP synthesis. However, when the ETC is damaged or inhibited—as seen in Long COVID—this process becomes inefficient and "leaky." Electrons can escape prematurely and react directly with molecular oxygen, forming highly reactive and damaging molecules known as reactive oxygen species (ROS), such as superoxide (O2•−).2
The excessive production of ROS initiates a cascade of oxidative damage. These molecules attack and damage critical cellular components, including lipids in the mitochondrial membranes, proteins of the ETC itself, and, critically, mitochondrial DNA (mtDNA).1 Damage to ETC proteins further impairs their function, leading to more electron leakage and even greater ROS production. Damage to mtDNA, which encodes essential components of the ETC, can lead to the synthesis of faulty proteins, permanently crippling the respiratory chain. This creates a devastating feedback loop where mitochondrial dysfunction generates ROS, and ROS, in turn, exacerbates mitochondrial dysfunction.33
This cycle is further amplified by a weakening of the body's antioxidant defenses. Studies have noted reduced levels of key antioxidants, such as glutathione, in Long COVID patients, diminishing the cell's capacity to neutralize ROS and mitigate oxidative damage.33 The net result is a state of severe and chronic redox imbalance. Indeed, studies analyzing immune cells from both Long COVID and ME/CFS patients have identified elevated oxidative stress as a core, shared molecular feature of these illnesses, linking it directly to functional impairments.35 This unmitigated oxidative stress not only damages mitochondria but also contributes to systemic inflammation and the overall pathology of the disease.
3.3 Disruption of Mitochondrial Homeostasis: Biogenesis, Dynamics, and Nuclear Communication
The pathology in Long COVID extends beyond damage to existing mitochondria to encompass a failure of the entire mitochondrial quality control and maintenance system. Healthy cells constantly renew their mitochondrial population through a coordinated system of biogenesis (creating new mitochondria), dynamics (fission and fusion), and mitophagy (removal of damaged mitochondria). In Long COVID, this homeostatic network is profoundly disrupted.
Evidence points to impaired mitochondrial biogenesis, the process responsible for generating new, healthy mitochondria to replace damaged ones and to adapt to increased energy demands.33 This process is largely controlled by the master regulator PGC-1α. The fact that therapeutic strategies are being explored to activate this pathway suggests that it is suppressed in Long COVID, hindering the cell's ability to repair and replenish its mitochondrial pool.33
Communication between the mitochondria and the cell nucleus, which is essential for coordinating cellular metabolism and responding to stress, is also compromised. The nucleus contains the vast majority of genes encoding mitochondrial proteins; therefore, a constant dialogue is required to match mitochondrial function with cellular needs. Studies have highlighted disruptions in these mitochondrial-nuclear signaling pathways in Long COVID, leading to a widespread dysregulation of metabolic processes and an amplification of the oxidative stress response.33
Finally, the physical morphology and interconnectivity of the mitochondrial network, governed by the processes of fission (division) and fusion (merging), are altered. Mitochondrial dynamics are critical for cellular health; fusion allows mitochondria to share components and dilute damage, while fission segregates damaged mitochondria for removal via mitophagy and facilitates the creation of new organelles.37 SARS-CoV-2 infection is known to disrupt this delicate balance, with in vitro studies showing that the Spike protein can induce mitochondrial fragmentation and swelling.19 Other research using sera from patients suggests a complex process that may begin with a stress-induced hyperfusion of the mitochondrial network, but upon prolonged exposure to the pathological factors in the sera, progresses to fragmentation and severe deterioration.41 The disruption of these dynamic processes prevents the efficient removal of damaged mitochondria and impairs the cell's ability to maintain a healthy, functional mitochondrial network, contributing to the accumulation of dysfunctional organelles and the overall bioenergetic collapse.
Section 4: Post-Exertional Malaise: The Clinical Manifestation of Bioenergetic Failure
Post-exertional malaise (PEM) is the defining and most disabling characteristic of Long COVID and ME/CFS. It is not simply fatigue but a systemic, multi-symptom exacerbation that occurs hours to days after an exertion that would have been previously well-tolerated.7 The synthesis of the evidence presented in the preceding sections allows for the construction of a comprehensive pathophysiological model of PEM, explaining it not as a mysterious or psychosomatic phenomenon, but as the direct, predictable, and logical collapse of a critically compromised cellular energy system.
4.1 The Bioenergetic Cliff: Why Exertion Triggers Systemic Collapse
At baseline, the skeletal muscle of an individual with Long COVID exists in a precarious state of chronic energy deficit. As established, the triad of viral, immune, and vascular insults has crippled the mitochondrial population, leading to severely diminished OXPHOS capacity and a forced reliance on inefficient anaerobic glycolysis.1 The cellular energy system operates with virtually no metabolic flexibility or reserve capacity. Healthy individuals can seamlessly ramp up mitochondrial ATP production by orders of magnitude to meet the demands of exercise, but in Long COVID, this capacity is lost. The system is locked into a low-output state.
Any form of exertion—be it physical, cognitive, or emotional—imposes an increased demand for ATP on this fragile system. The compromised mitochondria are fundamentally unable to increase their rate of aerobic respiration to meet this new demand. This acute mismatch between cellular energy demand and the system's maximum supply capacity is the initiating event of PEM.1 When demand exceeds the low ceiling of aerobic production, the cell is pushed over a "bioenergetic cliff," triggering a cascade of harmful downstream consequences. This model reframes PEM as a direct and logical consequence of a supply-demand mismatch in a system with no metabolic reserve. It is the predictable system failure that occurs when a critically damaged engine is pushed beyond its operational limits.
4.2 The Pathological Cascade of PEM
The failure to meet energy demand aerobically sets off a multi-system pathological cascade that accounts for the diverse symptoms and delayed onset of PEM.
Metabolic Shift and Acidosis: To generate any ATP at all, the muscle cell must massively upregulate anaerobic glycolysis. This pathway is fast but extremely inefficient, producing only 2 ATP per molecule of glucose compared to ~36 via OXPHOS. This desperate metabolic shift leads to the rapid accumulation of byproducts, particularly lactate, causing a drop in intracellular pH (acidosis). This acidosis contributes directly to muscle pain, impairs the function of contractile proteins, and exacerbates the sensation of fatigue.30
Oxidative Stress Surge: The struggling mitochondria, pushed beyond their functional capacity, leak an enormous number of electrons. This results in a massive burst of ROS, overwhelming the already depleted antioxidant defenses.33 This surge in oxidative stress causes widespread damage to cellular structures, further impairing mitochondrial function and activating inflammatory pathways.
Acute Myopathy and Tissue Damage: The combination of severe energy depletion (ATP crisis), intracellular acidosis, and the massive surge in oxidative stress is profoundly toxic to the muscle cell. This leads to acute cellular injury and death, which is directly observed as the fiber necrosis, inflammation, and increased amyloid deposition seen in post-exertion muscle biopsies.4 This is the direct evidence of exertion-induced tissue damage.
Worsening Mitochondrial Function: The metabolic stress of exertion inflicts further damage upon the already fragile mitochondrial population. This is evidenced by the significant drop in SDH activity observed in the day following PEM induction.4 This creates a dangerous feedback loop where each episode of PEM can cause a net loss of mitochondrial function, potentially lowering the patient's overall functional baseline and making them more susceptible to future crashes.
The characteristic delayed onset of PEM (often 24-72 hours post-exertion) can be explained by the timeline of these biological events.7 The initial exertion triggers the immediate bioenergetic crisis and metabolic shift. The subsequent and more prolonged symptoms are the clinical manifestation of the consequences of that crisis: the inflammatory response mounted by the immune system to clear the necrotic tissue, the time required for complex cellular repair processes to engage, and the slow process of clearing the accumulated metabolic waste and oxidative damage. The prolonged recovery time—lasting days, weeks, or even longer—reflects the severity of the damage inflicted and the profoundly impaired capacity of the body to generate the very energy needed for repair and recovery.
Section 5: A Broader Context: Pathophysiological Parallels with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)
The emergence of Long COVID on a global scale has provided an unprecedented opportunity to study post-infectious, energy-limiting illnesses. The striking similarities between the clinical presentation and underlying biology of Long COVID and Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS), a condition often triggered by other infections, strengthens the biological plausibility of the mechanisms described in this report. This comparison suggests that Long COVID is not an entirely novel entity but rather a prominent example of a common final pathway of disease that can be initiated by various triggers.
5.1 Shared Clinical Phenotypes
The clinical overlap between Long COVID and ME/CFS is extensive. A significant proportion of individuals with Long COVID, estimated to be around half in some prospective studies, meet the established diagnostic criteria for ME/CFS.44 The core symptom clusters are virtually identical, headlined by debilitating, persistent fatigue that is not alleviated by rest; cognitive dysfunction, often termed "brain fog"; and the hallmark feature of post-exertional malaise (PEM).36 Both conditions are multisystemic, with patients also reporting widespread pain (myalgia and arthralgia), unrefreshing sleep, and autonomic dysfunction such as orthostatic intolerance.36 This profound clinical similarity strongly implies a shared underlying pathophysiology.
5.2 Overlapping Biological Abnormalities
Research into the biological underpinnings of these conditions has revealed a remarkable convergence of findings, particularly in the domains of energy metabolism and exercise intolerance.
Impaired Oxygen Extraction: Invasive cardiopulmonary exercise testing has demonstrated that a central mechanism of exercise limitation in both Long COVID and ME/CFS is severely impaired systemic oxygen extraction and reduced skeletal muscle oxygen diffusion (DM).27 This shared finding points to a common peripheral, neuromuscular bottleneck that prevents oxygen from effectively reaching the mitochondria during exertion. This could be driven by microvascular pathology, autonomic dysregulation of blood flow, or intrinsic mitochondrial defects.27
Mitochondrial Dysfunction: Mitochondrial impairment is a central theme in the pathophysiology of both illnesses. Studies in ME/CFS have long reported abnormalities consistent with those now being found in Long COVID, including evidence of impaired ATP synthesis, an earlier shift to anaerobic metabolism during exercise, and elevated markers of oxidative and nitrosative stress.35
Immune Dysregulation: Both conditions are characterized by evidence of persistent immune dysregulation. This includes patterns of chronic inflammation, altered cytokine profiles, and signs of dysfunctional T-cell responses.47 The shared post-infectious nature of many ME/CFS cases and all Long COVID cases suggests that a dysregulated immune response to an initial pathogen is a key initiating event.
While the overlap is substantial, some studies suggest potential distinctions that may be related to disease duration or specific triggers. For example, one comparative study found that while both ME/CFS and Long COVID (termed Post-COVID Syndrome, PCS) patients had impaired mitochondrial function, particularly in Complex I of the ETC, the ME/CFS group exhibited more severe and progressed pathological structural changes to their mitochondria (e.g., smaller, more deformed organelles). In contrast, the dysfunction in the PCS group appeared to be more acutely functional, possibly reflecting a more direct and recent viral impact.45 This suggests that while the functional deficits may be similar, the long-term chronicity of ME/CFS may lead to more profound, cumulative structural damage over time.
The extensive parallels in clinical presentation, objective physiological dysfunction, and cellular pathology strongly suggest that Long COVID and ME/CFS represent points on a spectrum of post-infectious energy metabolism disorders. It appears that different infectious triggers (SARS-CoV-2, Epstein-Barr virus, enteroviruses, etc.) or even non-infectious insults can initiate a common final pathophysiological pathway characterized by a triad of immune dysregulation, endothelial/vascular dysfunction, and mitochondrial collapse. This reframes Long COVID not as a unique disease, but as a massive, globally observable cohort of a long-recognized but historically neglected class of illnesses, providing a critical opportunity to advance the understanding of the entire spectrum of post-exertional, energy-limiting diseases.
Section 6: Synthesis, Implications, and Future Trajectories
The comprehensive body of evidence reviewed in this report converges to form a coherent and compelling model of long-term mitochondrial dysfunction in the skeletal muscle of Long COVID patients. This model explains how an initial SARS-CoV-2 infection can trigger a self-perpetuating cycle of cellular injury and bioenergetic failure, leading directly to the profound fatigue and post-exertional malaise that characterize the condition. This synthesis has profound implications for how the disease is diagnosed, treated, and managed, and illuminates clear trajectories for future research.
6.1 A Unified Model of Mitochondrial Failure in Long COVID
The pathophysiology of mitochondrial dysfunction in Long COVID can be conceptualized as a multi-stage cascade:
Initiating Insults: The process begins with the "triad of insult" from the SARS-CoV-2 infection. This includes direct molecular disruption from circulating viral proteins, a "hit-and-run" cascade of immune-mediated damage initiated by a specific cytokine signature (TNF-α and IFN-γ) that transcriptionally suppresses mitochondrial genes, and widespread microvascular pathology (endothelialitis and fibrin amyloid microclots) that creates a state of chronic tissue hypoxia.
Systemic Consequences: These primary insults create a hostile systemic and local environment for the mitochondria. Chronic inflammation, potential autoimmunity, and persistent tissue hypoxia prevent cellular recovery and actively perpetuate damage.
Mitochondrial Collapse: The convergence of these pressures on the mitochondria leads to a complete system failure. This is characterized by a catastrophic breakdown in aerobic energy production (impaired TCA cycle and OXPHOS), a vicious cycle of excessive ROS production and oxidative stress, and a disruption of essential mitochondrial homeostasis and quality control systems (biogenesis, dynamics, and mitophagy).
Clinical Manifestation: This profound and persistent cellular energy crisis manifests clinically as the core symptoms of Long COVID. The chronic inability to produce sufficient ATP leads to debilitating fatigue, muscle weakness, and cognitive dysfunction. The system's extreme bioenergetic fragility and lack of reserve capacity mean that any increase in energy demand triggers the pathological cascade of PEM, resulting in acute tissue damage and a prolonged, multi-symptom crash.
6.2 Answering the Core Query: Direct vs. Indirect Impairment
The central question posed was whether SARS-CoV-2 directly impairs mitochondrial replication and energy production pathways. The evidence synthesized in this report indicates a nuanced answer: the virus instigates mitochondrial dysfunction through both direct and indirect mechanisms, with the indirect pathways being the primary drivers of the long-term, chronic nature of the condition.
Direct Impairment: There is clear in vitro evidence that SARS-CoV-2 proteins, particularly the Spike protein, can directly interact with and inhibit the function of mitochondrial ETC complexes, alter membrane potential, and disrupt mitochondrial morphology.15 The persistence of these proteins in the circulation of some patients suggests this direct mechanism may contribute to ongoing pathology.14
Indirect Impairment: However, the most compelling evidence for the chronicity and severity of the dysfunction points to powerful indirect mechanisms. The two most significant are:
Immune-Mediated Transcriptional Suppression: The initial acute-phase cytokine storm appears to trigger a lasting, possibly epigenetic, suppression of the nuclear genes required for building and maintaining the mitochondrial respiratory machinery. This "molecular memory" of the infection locks the cell into a low-energy state long after the virus is cleared.5
Hypoxia-Driven Metabolic Shift: The persistent microvascular disease and resulting tissue hypoxia create an insurmountable external constraint on mitochondrial function. Mitochondria cannot perform aerobic respiration without oxygen. This chronic oxygen supply crisis forces a pathological reliance on anaerobic metabolism and inflicts continuous damage via ischemia/reperfusion injury.24
Therefore, while direct viral protein interactions may initiate or contribute to the damage, it is the perpetuating cycles of immune dysregulation and vascular compromise that appear to be the dominant forces preventing mitochondrial recovery and driving the long-term disease state.
6.3 Implications for Diagnostics, Therapeutics, and Rehabilitation
This mechanistic understanding has critical implications for clinical practice:
Diagnostics: Standard clinical blood tests and imaging often fail to capture the underlying pathology of Long COVID. The findings strongly advocate for a shift towards functional and specialized diagnostics. This includes the use of invasive or non-invasive cardiopulmonary exercise testing to objectively measure impaired oxygen extraction, which appears to be a key physiological signature of the disease.27 Furthermore, there is an urgent need to develop and validate novel biomarkers that reflect the core pathologies, such as assays for fibrin amyloid microclots, markers of endothelial dysfunction (e.g., ANG-1/P-SEL), and panels measuring oxidative stress.23
Therapeutics: A single-target therapeutic approach is unlikely to succeed. The multifactorial nature of the disease necessitates multi-pronged treatment strategies that address the upstream drivers. This could include combinations of:
Antivirals for patients with evidence of persistent viral reservoirs.14
Anti-platelet/anticoagulant therapies and endothelial-stabilizing agents to address the microvascular pathology and improve tissue perfusion.24
Immunomodulatory agents to quell chronic inflammation and potentially reverse autoimmune processes.
Mitochondrial support therapies aimed at mitigating oxidative stress (e.g., antioxidants like Coenzyme Q10, N-acetylcysteine) and providing essential cofactors for energy metabolism (e.g., L-carnitine, B vitamins).2
Rehabilitation: The unequivocal evidence of severe, exercise-induced muscle necrosis and the worsening of mitochondrial function following exertion provides a powerful biological rationale against the use of rehabilitative approaches like graded exercise therapy (GET) for patients with PEM.4 Such approaches risk causing iatrogenic harm by repeatedly triggering the PEM cascade and potentially lowering a patient's functional baseline. Instead, clinical guidance should strongly emphasize energy management strategies, such as pacing, to help patients stay within their limited energy envelope and avoid inducing this harmful biological process.7
6.4 Future Research Trajectories
While significant progress has been made, critical questions remain. Future research should prioritize several key areas:
Longitudinal Studies: There is a need for longitudinal studies that perform serial muscle biopsies and functional assessments in the same patients over time to track the evolution of the muscle pathology and its response to interventions.
Biomarker Validation: Robust efforts are required to validate promising biomarkers (e.g., microclots, circulating PRDX3, venous succinate) in large, diverse patient cohorts to facilitate patient stratification and serve as objective endpoints in clinical trials.26
Mechanism of Transcriptional Suppression: Elucidating the precise epigenetic mechanisms (e.g., DNA methylation, histone modification) by which the initial cytokine storm leads to the long-term suppression of mitochondrial genes could reveal novel therapeutic targets for "reprogramming" the cell back to a healthy metabolic state.
Clinical Trials: Rigorous, randomized controlled trials are urgently needed to test the multi-pronged therapeutic strategies outlined above. These trials should use both clinical outcomes and objective biological markers to assess efficacy.
In conclusion, the profound fatigue and post-exertional malaise of Long COVID are rooted in a severe, multi-system pathology that converges on the mitochondrion. A complex interplay of direct viral insults, chronic immune dysregulation, and microvascular disease cripples the ability of skeletal muscle to produce energy, creating a state of extreme bioenergetic fragility. Addressing this cellular energy crisis will be the central challenge in developing effective treatments and restoring health to the millions living with this debilitating condition.
Acknowledgement
I acknowledge the use of Gemini AI in the preparation of this report. Specifically, it was used to: (1) brainstorm and refine the initial research questions; (2) assist in writing and debugging Python scripts for statistical analysis; and (3) help draft, paraphrase, and proofread sections of the final manuscript. I reviewed, edited, and assume full responsibility for all content.
Works cited
COVID-19 Toes and Other Skin Lesions During the Pandemic ..., accessed September 9, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC9647229/
Dermatological Manifestations of COVID-19: A Comprehensive Review | Published in Physician's Journal of Medicine, accessed September 9, 2025, https://www.physiciansjom.org/article/131900-dermatological-manifestations-of-covid-19-a-comprehensive-review
Understanding the role that 'COVID toe' has in recognizing the potential extent of COVID-19 infections: a case study - PMC, accessed September 9, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC7480604/
View of COVID toes: a unique cutaneous indicator of COVID-19 | The Southwest Respiratory and Critical Care Chronicles, accessed September 9, 2025, https://pulmonarychronicles.com/index.php/pulmonarychronicles/article/view/845/1777
Pseudo-Chilblains in Adult Patients with Confirmed COVID-19: A Systematic Review, accessed September 9, 2025, https://ijms.info/IJMS/article/view/1648/2229
Pernio (Chilblains), SARS-CoV-2, and COVID Toes Unified Through ..., accessed September 9, 2025, https://pubmed.ncbi.nlm.nih.gov/33714595/
Chilblains-Like Lesions in Pediatric Patients: A Review of Their Epidemiology, Etiology, Outcomes, and Treatment - Frontiers, accessed September 9, 2025, https://www.frontiersin.org/journals/pediatrics/articles/10.3389/fped.2022.904616/full
SARS‐CoV‐2 endothelial infection causes COVID‐19 chilblains: histopathological, immunohistochemical and ultrastructural study of seven paediatric cases, accessed September 9, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC7323219/
What are COVID Toes? - News-Medical.net, accessed September 9, 2025, https://www.news-medical.net/health/What-are-COVID-toes.aspx
Pseudo-Chilblains in Adult Patients with Confirmed COVID-19: A Systematic Review, accessed September 9, 2025, https://ijms.info/IJMS/article/view/1648/2228
Histopathology of persistent long COVID toe: A case report - PMC - PubMed Central, accessed September 9, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC9111773/
When interferon tiptoes through COVID-19: Pernio-like lesions and ..., accessed September 9, 2025, https://www.researchgate.net/publication/342306796_When_interferon_tiptoes_through_COVID-19_Pernio-like_lesions_and_their_prognostic_implications_during_SARS-CoV-2_infection
Histopathology of Persistent Long COVID Toe: A Case Report, accessed September 9, 2025, https://ammes.org/2022/04/13/histopathology-of-persistent-long-covid-toe-a-case-report/
“COVID toes”: A true viral phenomenon or a diagnosis without a leg to stand on? - PMC, accessed September 9, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC9213024/
Interferon Responses in “COVID Toes,” Footprints from SARS-CoV2 Infection, accessed September 9, 2025, https://wpp.med.wisc.edu/funded-project/interferon-responses-in-covid-toes/
Chilblain-like lesions onset during SARS-CoV-2 infection in a COVID-19-vaccinated adolescent: case report and review of literature - PMC, accessed September 9, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC9190458/
'COVID toes' were likely not caused by COVID-19, in most cases, accessed September 9, 2025, https://divisionofresearch.kaiserpermanente.org/covid-toes-not-caused-by-covid/
Population Characteristics, Symptoms, and Risk Factors of Idiopathic Chilblains: A Systematic Review, Meta-Analysis, and Meta-Regression - MDPI, accessed September 9, 2025, https://www.mdpi.com/2079-7737/11/11/1651
When interferon tiptoes through COVID-19: Pernio-like lesions and ..., accessed September 9, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC7303035/
New insights in COVID-19–associated chilblains: A comparative study with chilblain lupus erythematosus | Request PDF - ResearchGate, accessed September 9, 2025, https://www.researchgate.net/publication/342660807_New_insights_in_COVID-19-associated_chilblains_A_comparative_study_with_chilblain_lupus_erythematosus
A clinico-pathological description of COVID-19-induced chilblains ..., accessed September 9, 2025, https://dipot.ulb.ac.be/dspace/bitstream/2013/328198/3/COVIDtoes.pdf
Pernio (Chilblains), SARS-CoV-2, and COVID Toes Unified Through Cutaneous and Systemic Mechanisms - PubMed Central, accessed September 9, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC7826004/
(PDF) SARS-CoV-2 and Skin: The Pathologist's Point of View - ResearchGate, accessed September 9, 2025, https://www.researchgate.net/publication/352152618_SARS-CoV-2_and_Skin_The_Pathologist's_Point_of_View
SARS-CoV-2 Detection by Digital Polymerase Chain Reaction and ..., accessed September 9, 2025, https://pubmed.ncbi.nlm.nih.gov/37075721/
RNAscope in situ hybridization and RT-PCR for detection of SARS-CoV-2 in chilblain-like lesions: A clinical, laboratory and histopathological study - PubMed, accessed September 9, 2025, https://pubmed.ncbi.nlm.nih.gov/34989043/
Histopathology of persistent long COVID toe: A case report. | University of Kentucky College of Arts & Sciences, accessed September 9, 2025, https://english.as.uky.edu/bibcite/reference/28650
Vascular Alterations Following COVID-19 Infection: A Comprehensive Literature Review, accessed September 9, 2025, https://www.mdpi.com/2075-1729/14/5/545
Skin manifestations with COVID-19: the purple skin and toes that ..., accessed September 9, 2025, https://journals.cambridgemedia.com.au/wcetes/volume-40-number-2/skin-manifestations-covid-19-purple-skin-and-toes-you-are-seeing-may-not-be-deep-tissue-pressure-injury
Platelet and Monocyte Microvesicles as Potential Biomarkers of COVID-19 Severity: A Cross-Sectional Analysis - Annals of Laboratory Medicine, accessed September 9, 2025, https://www.annlabmed.org/journal/view.html?doi=10.3343/alm.2023.0395
The skin as a critical window in unveiling the pathophysiologic principles of COVID-19 - PMC - PubMed Central, accessed September 9, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC8298003/
Pernio (Chilblains), SARS-CoV-2, and COVID Toes Unified Through Cutaneous and Systemic Mechanisms - ResearchGate, accessed September 9, 2025, https://www.researchgate.net/publication/348685626_Pernio_Chilblains_SARS-CoV-2_and_COVID_Toes_Unified_Through_Cutaneous_and_Systemic_Mechanisms
Idiopathic chilblains and COVID toes. - European Journal of Pediatric Dermatology, accessed September 9, 2025, https://ejpd.com/index.php/journal/article/view/2216
Investigating the Link Between 'COVID Toes' and the Virus ..., accessed September 9, 2025, https://wpp.med.wisc.edu/featured-grantees/lisa-arkin-marie-singh-covid-toes/
Correlation between Type I Interferon Associated Factors and COVID-19 Severity - MDPI, accessed September 9, 2025, https://www.mdpi.com/1422-0067/23/18/10968
Type I interferon response and vascular alteration in chilblain-like lesions during the COVID-19 outbreak | Request PDF - ResearchGate, accessed September 9, 2025, https://www.researchgate.net/publication/355130987_Type_I_interferon_response_and_vascular_alteration_in_chilblain-like_lesions_during_the_COVID-19_outbreak
Are chilblains a skin expression of COVID‐19 microangiopathy? - PMC - PubMed Central, accessed September 9, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC7405401/
SARS-CoV-2 detection by digital polymerase chain reaction and immunohistochemistry in skin biopsies from 52 patients with different COVID-19-associated cutaneous phenotypes - Bohrium, accessed September 9, 2025, https://www.bohrium.com/paper-details/sars-cov-2-detection-by-digital-polymerase-chain-reaction-and-immunohistochemistry-in-skin-biopsies-from-52-patients-with-different-covid-19-associated-cutaneous-phenotypes/856118843892826113-12189
Discordant anti-SARS-CoV-2 spike protein and RNA staining in ..., accessed September 9, 2025, https://pubmed.ncbi.nlm.nih.gov/32895985/
COVID-Toes: The Clinical Correlation between Chilblains and SARS-CoV-2, accessed September 9, 2025, https://sma.org/abstracts/covid-toes/
Pathogenic Basis of Thromboinflammation and Endothelial Injury in COVID-19: Current Findings and Therapeutic Implications - MDPI, accessed September 9, 2025, https://www.mdpi.com/1422-0067/22/21/12081
'COVID toes' clinical trial underway at UW School of Medicine and Public Health, accessed September 9, 2025, https://www.med.wisc.edu/news/covid-toes-clinical-trial-underway/
Research reveals mechanisms behind “COVID toes” - BAD Patient Hub - Skin Health Info, accessed September 9, 2025, https://www.skinhealthinfo.org.uk/research-reveals-mechanisms-behind-covid-toes/
about — COVID HUMAN GENETIC EFFORT, accessed September 9, 2025, https://www.covidhge.com/about